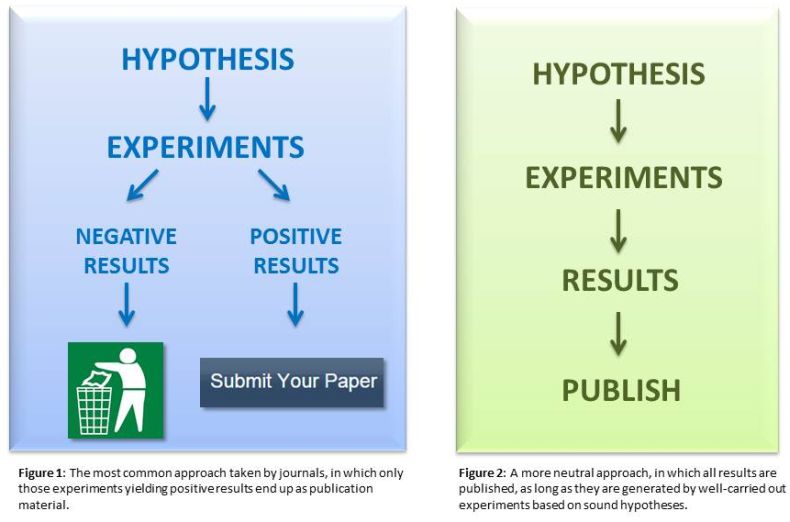
One of the intermediate stages of my final project.
Topic chosen: Cancer and DNA methylation
Specific question: Does loss of DNMT3A lead to changes in HOXA9 methylation that contribute to leukemogenesis?
How is this question novel and original? DNMT3A null mutations in acute myeloid leukemia were only discovered in 2010. To date, the majority of studies have only examined the presence of DNMT3A mutations in clinical leukemia samples, with few performing “add something” or “remove something” experiments. Those that did examine DNMT3A knockout’s effect in AML did not delve into the mechanisms through which this increased propensity to cancer might be occurring. This study will investigate a potential avenue through which DNMT3A exerts its leukemogenic function.
Hypothesis: I hypothesize that knockout of DNMT3A will lead to hypomethylation of the HOXA9 gene, which will in turn increase the cells’ propensity to develop into AML.
Evidence on which the hypothesis is based:
- DNMT3A is frequently mutated in acute myeloid leukemia (Yang et al., 2015)
- DNMT3A mutation is one of the first mutations to occur in DNMT3A-/- AML and increases likelihood of developing into cancer (Shlush et al., 2014)
- Homeobox-containing (HOX) genes were found to be hypomethylated in DNMT3A-/- cancers (Qu et al., 2014)
- HOXA9 overexpression has been implicated in AML development and poor prognosis (Collins and Hess, 2016)
Predictions: DNMT3A knockout will result in hypomethylation of the HOXA9 promoter, which will in turn lead to increased gene expression from the HOXA9 locus. This upregulation of HOXA9 will render cells predisposed towards AML.
Experimental approach to test prediction:
- Knockout of DNMT3A gene – catalytic domain, likely using CRISPR/Cas9 system or siRNA
- Methylation status of HOXA9 – measure via bisulfite conversions and pyrosequencing
- Protein expression, mRNA expression of HOXA9 – Western blot, RT-qPCR
- Xenograft or tumourigenesis assay in vitro to assess leukogenic capacity of cells
List of relevant primary and review articles read:
Yang, L., Rau, R., Goodell, M.A. (2015). DNMT3A in hematological malignancies. Nature Reviews Cancer. 15:152-165.
This review summarizes the research connecting mutations in the DNA methyltransferase 3A (DNMT3A) enzyme to blood malignancies such as acute myeloid leukemia (AML). This paper provided crucial information surrounding the structure of the DNMT3A enzyme, its catalytic activity, and the role it plays during development. The authors also examine the research implicating DNMT3A mutations in various blood malignancies of the myeloid and leukoid lineage, and propose a model for leukemia development in which DNMT3A acts to transform cells into a “pre-leukemic” state, and subsequent mutations in key proteins such as NPM1 result in different types of blood malignancies. This model greatly informed my own hypothesis and predictions for my research project.
Qu, Y. et al. (2014). Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 9(8):1108-1119.
This primary research study characterized the genome-wide methylation differences between cytogenically normal AML (CN-AML) cells and healthy control bone marrow cells. A key finding from this study was that homeodomain-containing (HOX) genes experienced the most significant changes in methylation, with hypomethylation occurring in genes including HOXA5 and HOXA9. While this study showed only a correlation between DNMT3A mutation and hypomethylation of homeobox genes, it provides an intriguing potential mechanism through which DNMT3A may initiate cancer.
Collins, C.T., Hess, J.L. (2016). Role of HOXA9 in leukemia: dysregulation, cofactors, and essential targets. Oncogene. 35(9):1090-8.
This review summarizes the role of the homeobox family protein HOXA9 in leukemia. HOXA9 is a transcription factor that plays several important roles in development, specifically in the expansion of hematopoietic stem cells (HSCs). The molecular mechanisms through which HOXA9 can induce leukemia are not yet understood, and it is hypothesized that several additional upstream genetic alterations leading to overexpression of HOXA9 have yet to be identified. Given that HOXA9 has also been found to be hypomethylated in DNMT3A-null cancers, this paper brought up the question of whether loss of DNMT3A and HOXA9 hypomethylation are connected to the gene’s overexpression and leukemogenic activity.
Potential ways to make your question known to the public at large:
- Biggest thing that people need to understand with cancer therapy and research is the heterogeneity of cancers – no two tumours are identical, which makes treatment especially challenging
- Need to explain concept of DNA methylation – enzyme (small molecular “machine”) adds a tag to DNA that turns it on or off
- Also emphasize the prevalence of AML – most common in older adults (65+) but still does affect younger people, has a poor prognosis (1 in 4 5-year survival rate)