Triggering event for cell dedifferentiation in Physcomitrella patens
Layman’s summary:
Stem cell research is important to many people because of its applications in medicine and disease therapy. However, understanding how to use stem cells in medical treatments is just one side of stem cell research. Another equally important facet to study is how stem cells are maintained and controlled in other living systems. Bryophytes—or mosses—are a unique group of plants that are able to turn any cell in their bodies into an entirely new plant. This ability is not found in any other organism, and how they are able to do this is not entirely known yet. Thus, in this study, I plan to investigate what events trigger cell dedifferentiation that leads to the regrowth of entire organisms from single leaf cells. Specifically, I will be testing whether cells innately contain molecules that prevent each other from degenerating back into stem cells. I will do this by removing cell contents from in-tact leaf cells and observing whether I can induce new plants to grow from the remaining leaf cells. Obtaining a holistic view of stem cell control and maintenance is vital in stem cell research, and it is my hope that understanding the stem-cell like qualities of bryophytes will lead to better understanding of stem cells in human models as well.
Introduction
Cell dedifferentiation is an event that is extremely rare in nature. Although some organisms are able to regenerate—for example, geckos can regrow tails (Alibardi 2009)—the ability to revert cells of a determined fate into one with totipotency is extremely uncommon. Plants can occasionally be induced by hormone baths to produce pluripotent ‘calli’ from cambium tissue (Xuand Hunag 2014), and some species can regenerate apical meristems when they are cut off (Xu et al 2006), but all of these examples involve the degeneration of a particular group of cells. That is, they already possess a pre-designated collection of cells able to regenerate structures, whereas most other cells will not have this ability. For instance, geckos cannot regrow tails using skin cells and plants cannot generally regrow roots using leaf cells. Thus, the ability for an organism to strip any cell of its identity and return it to an embryonic state is extremely rare.
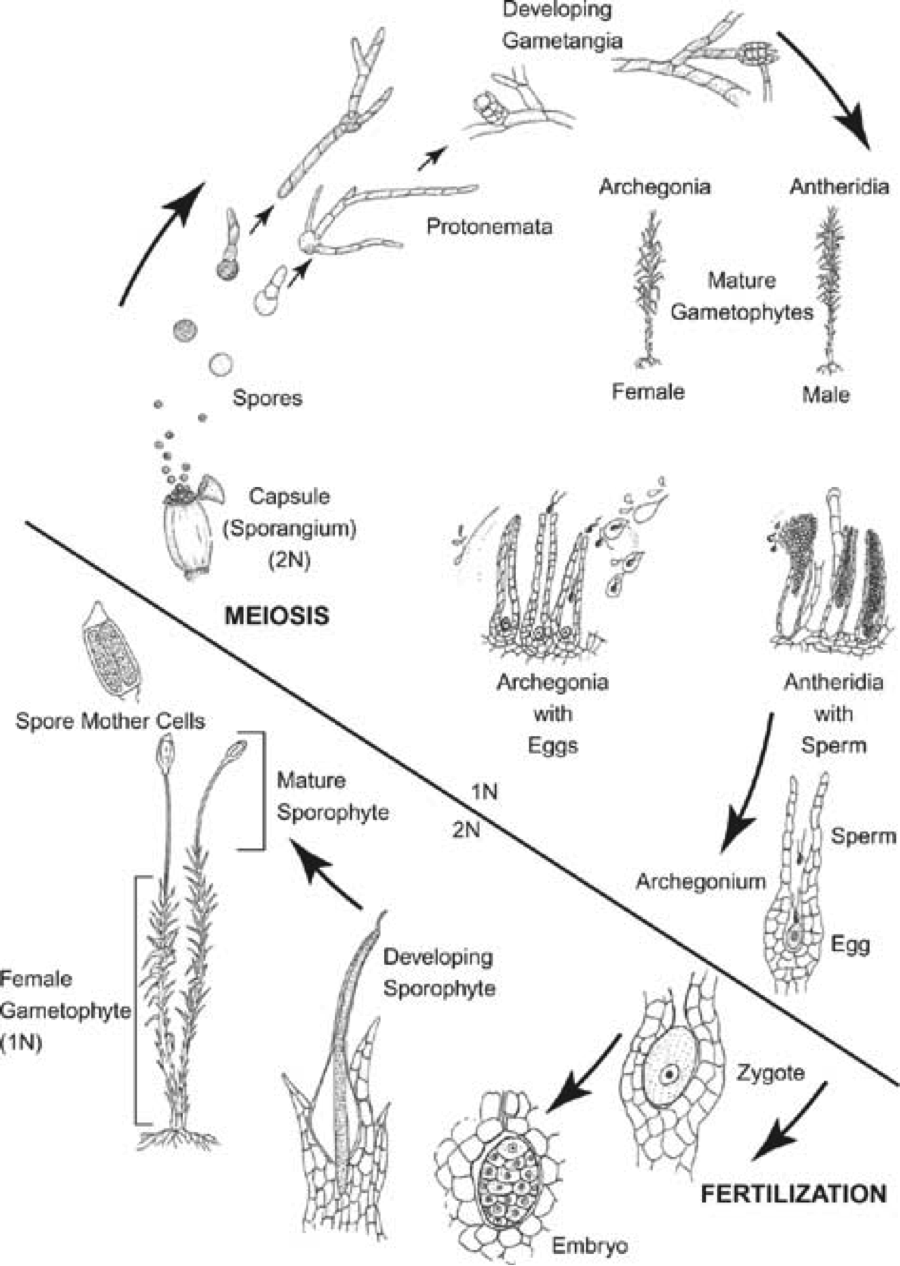
Interestingly, bryophytes are one of the few groups of organisms that can fully dedifferentiate any cell in their body to re-grow an entirely new entity. The ability for bryophytes to dedifferentiate and propagate itself via fragmentation is a trait that has been observed for a long time. The leafy gametophyte, rhizoids, and sporophyte have all been reported to dedifferentiate when excised from the parental plant, making it one of the few organisms that are not limited by particular stem cell ‘niches’ during regeneration (Giles 1971, Westerdijk 1907, Von Wettstein 1924, von Maltzahn 1959). Additionally, it seems that bryophytes do not require any exogenous signals to begin dedifferentiation, and that plant fragments will only begin to dedifferentiate when they are removed from the main structure (Giles 1971). Excised fragments will start to show dedifferentiation in certain cells very quickly—as soon as 24h—and cells destined for dedifferentiation will begin to grow and increase the amount of chloroplasts it has. Then, the cell will asymmetrically divide to form a basal cell and apical cell: the latter of which is nearly identical to young chloronema; a pluripotent filament traditionally seen emerging from spores (Giles 1971, Ishikawa et al. 2011, Sakakibara et al 2014). The apical cell will then act as a new chloronema filament and eventually give rise to an entirely new plant.
[For more information about the bryophyte life cycle and accompanying terminology, see posts in “Project” section on blog]
Unfortunately, the processes and mechanisms surrounding dedifferentiation in bryophytes is complicated and fairly inconsistent across species. Some species like Funaria will produce numerous apical chloronema cells across entire excised leaves, whereas species like Physcomitrella patens will produce protonema from just cells bordering the detached boundary of the leaf (Giles 1971, Westerdijk 1907). Others yet do not differentiate at all: the genus Dawsonia will increase chloroplast number and cell size but ultimately never form functional protonema (Von Wettstein 1924).
Not all cells are created equal either. Often, there is a gradient of most differentiable tissue to least, such that sporophytic tissue will dedifferentiate more readily near the apex whereas protonema will dedifferentiate better near the basal area than the tip (Von Wettstein 1924). Furthermore, the fact that sporophytic tissue and gametophytic tissue (which are diploid and haploid, respectively) are able to give rise to the same kind of chloronema is rather strange. This may be due to the fact that all gametophytic tissue in bryophytes are arrested in the G2 phase of the cell cycle, which means they all possess a duplicated (but still haploid) genome (Ishikawa et al 2011). This is in contrast to all other land plants and animals, whose cells are usually stopped in the G1 phase (Ishikawa et al 2011). Indeed, the dedifferentiation process of bryophytes is complex and seemingly difficult to understand.
There have been a handful of papers investigating the cellular changes associated with dedifferentiation in mosses. Ishikawa et al (2011) is a particularly notable example. Not only were they able to establish the duplicated state of the gametophytic genome, but also identified two essential proteins involved in triggering the dedifferentiation event in P. patens: CDKA and CDKD (cyclin-dependent kinases). It appeared that CDKA was an activator for CDKD, and while CDKA was found to be present in all cells at all times, CDKD was only transcribed in cells at detachment sites. Sakakibara et al (2014) has also contributed to the knowledge about bryophyte dedifferentiation by identifying a homolog of stem cell regulators in flowering plants (PpWOX13L) that is essential for the initiation of growth prior to the asymmetrical division of pre-chloronema. However, although both of these studies offer insight into what pathways are active post-trigger, the actual triggering mechanism for dedifferentiation in mosses still remains a mystery.
There have been some theories as to how cells are able to trigger dedifferentiation in excised plant fragments. Von Maltzahn (1959) suggested that leaves (and other tissue) receive signals form the apex that specify not to dedifferentiate, and that when leaves are removed from the main plant, the lack of this signal may result in spontaneous activation of CDKA/CDKD or PpWOX13L pathways. This idea of an innate ‘instability’ in bryophyte systems can be supported by similar systems seen in angiosperms. Higher plants are able to react to injury by using auxin as a concentration-dependent signal. Normally, there is a constant auxin flow from the apical tip to the peripheral tissues, but when tissues are damaged along this flow it slows or blocks the flow of auxin. This results in a build up of auxin on the basal side and a deficiency of auxin on the apical side, which initiates transcription to begin healing and regeneration (Asahina et al 2011, Read and Ross 2011). One can imagine how bryophytes may use a similar system to signal to leaves when dedifferentiation is necessary.
Other possible triggers for dedifferentiation include the use of external cues or hormones. The protonemata in bryophytes use hormones to communicate or detect each other by excreting substances into the environment. It is then the concentration of these hormones that permit or restrict bud formation. It is possible that excised leaves may be triggered to dedifferentiate because of similar reasons; namely because of the build-up of excreted residues that self-promote dedifferentiation when in contact with soil or media. In addition, previous studies have shown that light can have an inductive effect on chloronema (Giles 1967, Maltzahn 1968) so it would not be out of the question for light to play a role in dedifferentiation activation.
Finally, it is also possible that wounding may directly initiate the transcription process for cell dedifferentiation by triggering the transcription for wounding factors. This would be in contrast to the polarity theory, in which there is an absence of factors rather than the addition of them. Perhaps exposure of cell cytoplasm to differential osmotic conditions causes differential gene expression, leading to cell dedifferentiation. This model would explain why some plants might show ubiquitous cell dedifferentiation and some only show it on cells bordering cut sites: The leaves removed from certain species may be more delicate than others, and are thus more prone to damage than others.
Thus, despite our increasing knowledge of bryophyte dedifferentiation, we have yet to determine how the process is actually triggered: a question that I plan to address.
I hypothesize that there is the presence of some ‘constant’ signal in each cell that stabilizes neighbouring cells. Consequently, I propose that by removing the cytoplasm of neighbouring cells, I can induce cell dedifferentiation due to the removal of these ‘stabilizing’ signals.
Significance
Stem cell research is important for a variety of reasons. In medicine, there is hope that stem cell therapy can cure diseases or regrow structures to help both humans and animals. Thus, the side of stem cell research the general public hears about predominantly revolves around direct human application. However, there are many avenues of study that surround stem cell research that are equally important.
It is important to understand how stem cells function and how they stay totipotent because that information might help us understand how to make treatment more effective. Additionally, learning about how other systems handle stem cells may lead to analogous pathways within our own bodies. Other applications of cell totipotency can be used in farming techniques. Being able to master cell dedifferentiation to create cloned strains of certain foods may be beneficial to many farmers and save time and money. Clearly, there are many reasons why we would want to learn more about stem cells. Bryophytes give a unique perspective into stem cell maintenance because they are able to dedifferentiate every single cell in their body to become totipotent. Thus, understanding the triggering events and pathways involved in cell dedifferentiation may lead to better understanding of how to reverse or halt cell differentiation in other systems. In summary, the pieces of information gained from this experiment can eventually be applied to the direct problems that humans face.
Experimental Approach:
For my experiment, I plan to remove the cytoplasm of leaf cells from in-tact gametophytes. I chose to use the species Physcomitrella patens because of its historical use in bryophyte research. It is commonly used in gene studies because homologus recombination in this system is fairly simple, and reproducing clones is easy due to its ability to regenerate by fragmentation. Additionally, since Physcomitrella patens is part of the class Bryophyta, it generally does not possess any ‘special’ features associated with some other classes of bryophytes. Class Bryophyta is often referred to as the “Have-nots” of the bryophyte world because they are used as the ‘null’ comparison for other traits specific to other classes. Thus, P. patens can provide a simple, effective model to broadly represent bryophytic characteristics.
The use of the reporter genes used in my experimental was inspired by the system created by Shaefer (1994) and Ishikawa et al (2014). All protocols have been successfully used in other experiments.
Culturing of P. patens
First, I will obtain a line of P. patens similar to the ones found in Ishikawa et al. (2011). Preferably, I would like to use the ProCYCD;1:NLS-GFP- GUS #263 P. patens line in their paper, which used the polyethylene glycol–mediated transformation system created by Nishiyama et al (2000) and Shaefer (1994) to introduce a GFP signal to the CDKD protein. If obtaining this strain is not possible, then I will simply re-create this strain from WT P. patens using the protocols found in the Ishikawa et al (2011) and Nishiyama et al (2000) papers. Cultures will be kept on BCDAT media, which is used for cultivation of protonema and gametophores (Ishikawa 2011, Hiwatashi and Hasebe 2004). I have attached a copy of the BCDAT recipe in the supplemental information.
Ten stock plates of P. patens will be grown in case of error. They will be incubated at 250 C in continuous white light using BCDAT media (Nishiyama et al 2000) and transplanted every 8 weeks to ensure optimal health. When growing experimental gametophytes, protonema will be isolated from stock plates and rinsed with sterile BCDAT liquid media three times before placing on a layer of autoclaved cellophane, which will then be placed on solid BCDAT media. Rinsing with liquid media will minimize contamination from bacteria and fungus, whereas the cellophane layer will prevent new protonema from growing into the media and consequently make isolation of plants easier. Experimental plates will be cultivated for 4 weeks at 250 C to produce gametophytic shoots (Ishikawa et al 2011), which will then be removed with tweezers, rinsed 3 times in 0.22um filter-sterilized water, and placed horizontally on cellophane on BCDAT plates.
Treatments
All treatments will begin with a plate of freshly inoculated P. patens as described above. There will be 14 plates total with a single gametophyte on each. Ten of these gametophytes will be subject to all four treatments on different leaves to account for differences in individuals or media. The remaining four gametophytes will have 3 replicates of each type of treatment to ensure treatments are not affecting the results of each other. Treatments will be randomized in position along a single gametophyte so as to prevent any leaf-age effects. Leaves for treatment will be chosen based on how flat against the media they are: leaves that lie flat on the cellophane will be preferentially chosen to ensure any budding protonema will have sufficient nutrients.
Cytoplasm will be removed by glass micropipettes stretched out to have a tip with a 0.2um diameter (Mackler 1992). Originally, the extraction of cell contents was done on hippocampal neuron cells but as far as I am aware, such procedures have also been done on plant cells without issue. The cells of bryophytes are very delicate so the integrity of the cell wall should not pose a problem.
- No-cytoplasm treatment
Using the micropipette, an entire row of cells across the broadest part of the leaf will have their cytoplasm removed. The removal of the cytoplasm will be done by aspirating the contents out under observation via a 10X dissecting scope and visual examination of the cells will confirm the absence of cytoplasmic contents. This will be done for 10 leaves on 10 different plants.
- Puncture-control (Negative control 1)
Using the same micropipettes and procedures as in treatment (1), cells will be punctured but not aspirated.
- Excise-control (Positive control)
Using a razor blade, single leaves will be cut at the broadest part and moved 1cm away from the main shoot using sterilized tweezers. We will be careful not to damage the cells as we are moving the leaf fragment.
- In-tact control (Negative control 2)
Leaves will remain in-tact and un-punctured on the main stem.
Observation
Using a light microscope, I will make and record observations at 1h, 12h, 24h, 48h, and 2 days. The following traits will be noted:
- chloroplast count (with counts from surrounding cells for comparison)
- Budding or abnormal growth (made between time points)
- Cell division (the formation of the apical chloronemal cell)
Additionally, I will also look at GFP fluorescence under UV light to evaluate CDKD expression.
Finally, I will look at total protonemal growth after 2 days.
Possible Results:
No-cytoplasm treatments:
If my hypothesis is correct, I expect to see cells growing into protonema on the ‘No-cytoplasm’ treatments. The cells bordering the row of no-cytoplasm cells are expected to be the ones giving rise to chloronema. If cells on both sides grow chloronema, it would suggest that each cell has the same stabilizing signal, and that there is no polarity regarding this signal. However, if there were some kind of polarity (for example, if the signal is being excreted by the apical growth cell), then one would expect only the cells apical of the no-cytoplasm divide to dedifferentiate into chloronema. Another possible result would be for the no-cytoplasm treatments to lack dedifferentiation in any cell at all. These results would suggest that there is no ‘stabilizing’ signal, but that dedifferentiation is likely triggered by differential gene expression in response to cell damage. With no cytoplasm (and thus no transcriptional machinery), there would be no chance for cells to express wounding signals. If this result occurred, it would have to be compared to the puncture-controls to see if injury alone can initiate cell dedifferentiation.
In terms of CDKD-GFP expression, I would expect the cells along the line of no-cytoplasm cells to show high expression at 24h and 48h (Ishikawa et al 2011), and for this expression to coincide with chloronema formation. An unexpected result would be if CDKD-GFP expression and chloronema formation were not displayed together: this would suggest that either CDKD activation is not exclusive to dedifferentiation, or that it may not be necessary for dedifferentiation.
Puncture-control treatments:
Assuming the puncture-control cells did not lose too much cytoplasm and that they retained the ability to repair themselves, I would expect to see no chloronemal growth. However, it is possible that the puncture would be too large and result in enough loss of cytoplasm to initiate cell dedifferentiation on neighbouring cells. I would attempt to observe any gradient-like changes that occur in the no-cytoplasm, puncture control, and complete excision treatments to see if the puncture control would have less response than the no-cyotplasm and complete excision controls. Additionally, I would use visual examination to observe how damaging the puncture is. If it were evident that the puncture control had lost a substantial amount of cytoplasm, then I would have to find a new way to remove the cytoplasm while minimizing cell damage.
The expected result would be to see CDKD-GFP expression in the cells neighbouring the punctured cells. If this were the case, you would also expect protonemal growth from the neighbouring cells. An interesting result would be if CDKD-GFP expression existed in the punctured cells more than the neighbouring cells. It would suggest that it is physical damage that directly initiates cell dedifferentiation. This would be compared with possible protonemal growth to see if it corresponds with the increase in CDKD-GFP expression. CDKD-GFP expression in the punctured cells would also suggest that removing the cytoplasm of damaged cells would prevent dedifferentiation in leaves as a whole. This is because CDKD-GFP expression in punctured cells would mean differential gene expression in only damaged cells, and that dedifferentiation is not due to the loss of neighbouring ‘suppressors’ or some kind of polarity signal. Thus, one would expect that if the punctured cells showed CDKD-GFP expression, then the no-cytoplasm cells would not show dedifferentiation at all.
Thus, if CDKD-GFP expression were found in the punctured cells, one would expect no CDKD-GFP expression or protonemal formation in the no-cytoplasm treatment because there would be no damage done to the neighbouring cells. This is in contrast to the possibility where no CDKD-GFP expression would be seen in the punctured cells but high expression in the neighbouring cells, which would support my original hypothesis regarding the presence of a ‘stabilizing’ factor.
Excise-control treatments
I am expecting the excise-control to show protonemal growth from the cut boundary and CDKD expression in those cells as well. This treatment has already been tested successfully by Ishikawa et al (2011) under nearly identical conditions; so any deviations from this result would suggest we have done something wrong. The only difference between Ishikawa et al (2011)’s treatment and my experiment is the presence of the whole-gametophyte 1cm away from the excised leaf. In the Ishikawa et al (2011) experiment, they transferred the excised leaf onto a different plate. Thus, if our results differ from theirs, it would suggest there is some signal from the main shoot that plays a part in controlling dedifferentiation. This signal could be cytoplasmic (which may diffuse across the agar) or exogenous (such as the hormones found excreted by protonema). Alternatively, there may be multiple pathways in activating cell dedifferentiation and there would need to be more investigation as to why certain pathways are activated at certain times.
In-tact treatments
It is expected that these leaves will not show any dedifferentiation because they were not wounded in any way. Most literature has stated that attached leaves will not grow protonema, but there have been a few observations of this happening. It is unclear why this is the case, but I suspect it might be because of damage done to the leaves when they are placed on agar during the whole-gametophyte transferring process. Another possibility would be that there may be hormonal signals constantly excreted by the main shoot that signal dedifferentiation, and that when the plant is erect, these signals naturally dissipate into the air or are washed away by rain. However, when plants are placed horizontally and are in contact with solid substrate, these signals accumulate around the leaves and signal dedifferentiation.
If the scenario described above occurs, I would suspect that there are multiple pathways to dedifferentiation because the exogenous hormone accumulation theory would not explain why only cells bordering excised leaves would sprout protonema. If the leaf were excreting a hormone that accumulates around itself to induce dedifferentiation, one would expect the centre leaf cells to be most prone to dedifferentiation because it would have the highest concentration of these accumulated hormones.