
Simcha Srebnik
Associate Professor, Chemical and Biological Engineering, UBC
Member, Institute of Applied Mathematics
PhD, University of California, Berkeley, 1998
BSc Chemical Engineering, University of Toledo, 1994
Room: CHBE 419
Tel: 604.827.4814
Email: simcha.srebnik@ubc.ca
Research Openings
Research in my group focuses on developing simple physical models to describe molecular behaviour.
!!! An opening for a graduate student available to start thesis studies at UBC IMMEDIATELY on the following topic: The use of molecular dynamics simulation to identify structural and surface properties of a capillary that will prevent cavitation and maintain a stable liquid state of water !!!
In addition, I am actively seeking undergraduate students for specific projects in the following two areas:
I. Protein Structure and Folding
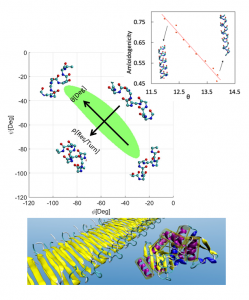 Using data mining and deep learning algorithms, combined with simple physical models, we advance our understanding of protein structure, folding, aggregation, and misfolding dynamics. Amyloids are one example where misfolding of a protein secondary structure (usually involving a transition from helical to sheet structures) can propagate into malignant superstructures. A simple physical interpretation of the dihedral angles representing alpha helical conformations in proteins allowed us to correlate amyloidal helices with their angular pitch, q. We are currently developing dynamic models to further our understanding of protein folding and misfolding.III
Using data mining and deep learning algorithms, combined with simple physical models, we advance our understanding of protein structure, folding, aggregation, and misfolding dynamics. Amyloids are one example where misfolding of a protein secondary structure (usually involving a transition from helical to sheet structures) can propagate into malignant superstructures. A simple physical interpretation of the dihedral angles representing alpha helical conformations in proteins allowed us to correlate amyloidal helices with their angular pitch, q. We are currently developing dynamic models to further our understanding of protein folding and misfolding.III
- A position is open for an undergraduate student to use statistical and machine learning techniques to analyze the protein data bank for structural descriptors of amyloidal polypeptide sequences.
- A position is open for an undergraduate student to develop a simple model for mechanisms of helix-to-beta sheet transition
II. Anion exchange membrane fuel cells (AEMFCs)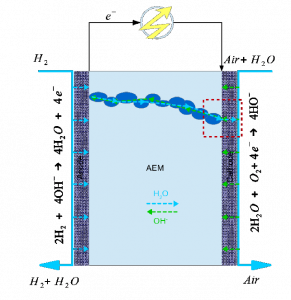
Rapid degradation of AEMFCs occurs due to the consumption of water at the cathode. At critically low water levels, hydroxide anions react with the cationic functional groups in the anion exchange membrane (AEM), limiting the conductivity of the AEM. At critically low water levels, we found new hydrated structures of two hydroxide anions at close proximity, bridged by water molecules. These surprising structures are very stable and have high activity.
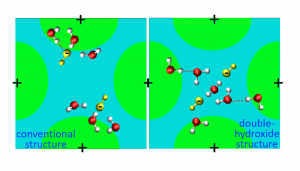
A position is open for an undergraduate student to further our understanding of AEM behaviour using advanced molecular simulation techniques.