Assignment 1:3 : Three Definitions
INTRODUCTION:
The focus of this assignment is to define relatively complex terms in three levels of detail for a non-technical audience. This document provides the parenthetical, sentence, and expanded definitions of the term “cystic fibrosis”, commonly used in the field of pulmonary research. I was first introduced to the disease during my participation in a research study at the Vancouver General Hospital.
TERM:
Cystic fibrosis
PARENTHETICAL DEFINITION:
Cystic fibrosis (a genetic disease) causes the body to produce mucus that is extremely thick and sticky.
SENTENCE DEFINITION:
Cystic fibrosis is a common genetic disease in which a mutated gene causes glands in the body to produce thick and sticky mucus in the respiratory tract, gastrointestinal tract, and certain reproductive organs.
EXPANDED DEFINITION:
What is Cystic Fibrosis?
Cystic Fibrosis is a progressively fatal genetic disease that causes exocrine glands to secrete thick and sticky mucus that causes infections, inflammation, blockages, indigestion, and malnutrition (Davies, Alton, & Bush, 2007). CF mainly affects the lungs, but can also affect other mucus-secreting body parts including the pancreas, reproductive organs, liver, and sinuses (Davies et al., 2007). The disease occurs when a person inherits two defective copies of the gene responsible for CF.
What is the history of Cystic Fibrosis?
Some of the earliest recorded cases (1595) of Cystic Fibrosis were marked by signs of a damaged pancreas and abnormally salty skin (Nick, 2012). It was first named in 1938 by American Pathologist Dr. Dorothy Andersen as “cystic fibrosis of the pancreas” after examining the autopsies of malnourished children (Nick, 2012). In the same year it was distinguished from celiac disease (Davis, 2006). In 1989 scientists discovered the gene responsible for the disease (CFTR) and sequenced its code (Nick, 2012). Currently, no cure for the disease has been discovered, but ongoing research is improving morbidity and mortality rates of CF (Davies et al., 2007).
What are the symptoms of Cystic Fibrosis?
Symptoms of CF vary with age and severity of disease. The most identifiable diagnosis is very salty-tasting skin, followed by persistent coughing at times with phlegm. Build-up of mucus in the lungs often leads to infections including pneumonia or bronchitis. Patients may also experience wheezing, shortness of breath, or nail clubbing (enlargement of fingertips, curving of nails around fingertips). Due to a weakened gastrointestinal system, there may be signs of poor growth or weight gain in spite of a good appetite, and difficulty with bowel movements. (Davies et al., 2007)
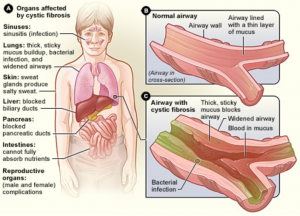
Figure 1. Areas of the body that show symptoms of Cystic Fibrosis. Airways that are clogged by thick, sticky mucus may be also infected.
Source: https://en.wikipedia.org/wiki/Cystic_fibrosis
[Accessed Sept 21, 2019]
What are the treatments for CF?
At the moment, there is no cure for CF but there are a number of treatments to help alleviate the problems caused by the disease. There are medicines that thin mucus and widen airways, and antibiotics that prevent and treat chest infections. Other medications and special diets help with food and nutrient absorption. Breathing techniques including forceful exhalation, and chest percussion help with expelling mucus (Figure 2). (“Cystic fibrosis”, 2019)
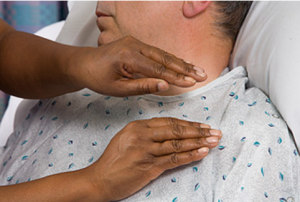
Figure 2. Chest percussion involves clapping above lobes of the chest with a cupped hand. The movement causes vibrations that help expel mucus.
Source: http://downloads.lww.com/wolterskluwer_vitalstream_com/sample-content/9780781788786_Craven/samples/mod09/topic10a/text.html
[Accessed Sept 21, 2019]
What are some misconceptions about CF?
Cystic Fibrosis is not selective to only Caucasians, but it may appear so because it is most common in that population. (Nick, 2012). Like many diseases, CF is not contagious; it needs to be inherited from both parents. However, even when both parents are carriers of the disease, there is only a 25% chance that their offspring will have CF. People affected by CF usually live to adulthood with an average life expectancy of 37 (“Myths about cystic fibrosis”, 2016).
WORKS CITED
Cystic fibrosis. (2019). Retrieved from https://www.nhsinform.scot/illnesses-and-conditions/lungs-and-airways/cystic-fibrosis
Davies, J. C., Alton, E. W., & Bush, A. (2007). Cystic fibrosis. BMJ (Clinical research ed.), 335(7632), 1255–1259. doi:10.1136/bmj.39391.713229.AD. Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2137053/
Davis, P. B. (2006). Cystic fibrosis since 1938. American Journal of Respiratory and Critical Care Medicine, 173(5), 475-482. doi:10.1164/rccm.200505-840OE. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/16126935
Myths about cystic fibrosis. (2016). Retrieved from https://www.aruma.com.au/about-us/blog/myths-about-cystic-fibrosis/
Nick, J. A. (2012). Cystic fibrosis: History. Retrieved from https://www.nationaljewish.org/conditions/cystic-fibrosis-cf/history
Leave a Reply